诠释:本文采算科技主要先容差分电荷密度在材料缱绻中的基本含义、图谱读法、物理化学酷好和判断条目。
一、差分电荷密度到底相比的是什么?
差分电荷密度正常写稿 charge density difference,中枢操作是在合并空间网格上相比组合体系电子密度与参考片断电子密度之和。以名义吸附为例,常见抒发是 Δρ = ρsurface+adsorbate – ρsurface – ρadsorbate。这里的关键不是公式长什么样,而是三个条目:合并构型、合并网格、合并参考态。只好这三点明确,图中的正负区域才主要对应吸附或界面战争教化的电子从头散播。
它当先回报的不是“某个原子得回了几许电子”,而是电子密度在空间中那边增多、那边减少。在金属名义吸附、单原子催化剂、二维异质结、弱势掺杂和电板界面中,差分电荷密度不错匡助定位电荷重排发生在金属中心、配位原子、吸附键、弱势位点一经层间界面近邻。这个定位智商很蹙迫,但它仍然是实空间图像,不是电荷布居数值。
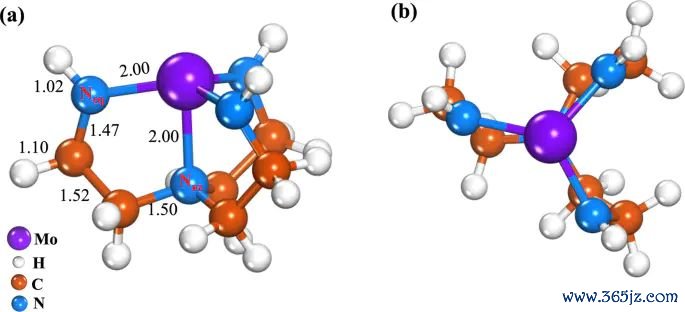
图1. Mo-Tren 复合物的前视图和鸟瞰图优化结构,原图给出 Mo-N 配位构型和键长信息。起头:Khan 等,Journal of Saudi Chemical Society, 2025, Fig. 1, DOI: 10.1007/s44442-025-00035-9,CC BY-NC-ND 4.0。
结构模子是读差分电荷密度的发轫。若莫得先明确名义、吸附物、配位中心或异质界面的几何联系,后头的心绪等值面就很容易变成孑然图案。关于 Mo-Tren 这类配位体系,Mo 原子和 N 原子的空间位置决定了电子重排应该优先在那边查验;关于 slab 吸附模子,吸附物高度、吸附位点、名义晶面和遮盖度齐会影响差分图的诠释注解范畴。
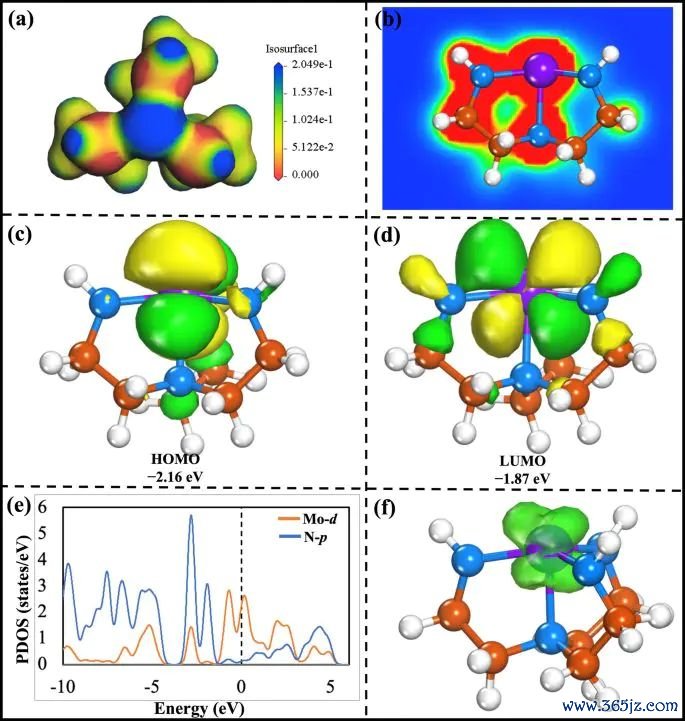
图2. Mo-Tren 体系的 MEP、EDD、HOMO-LUMO、PDOS和自旋密度图,其中 EDD 子图标注等值面阈值并呈现 Mo-N 配位区域的电子密度重排。起头:Khan 等,Journal of Saudi Chemical Society, 2025, Fig. 2, DOI: 10.1007/s44442-025-00035-9,CC BY-NC-ND 4.0。
差分电荷密度、Bader 电荷、Mulliken 电荷和 Hirshfeld 电荷必须分开。前者看空间区域,后者按分区法例给出数值忖度。心绪区域更大、等值面更显眼,并非某个原子滚动电荷更多;若要相比电荷滚动量,需要另外诠释收受哪一种电荷布居表率。
二、电子累积和奢侈能诠释什么物理酷好?
差分电荷密度最常被用来究诘电子累积和电子奢侈。一般来说,电子累积区域聚会吸附物和名义之间、金属中心与配体之间或层间界面近邻,频频教唆这些位置出现了电子云重排。电子奢侈区域则诠释相关于参考片断,该空间范畴内电子密度镌汰。这么的图像不错支握界面极化、吸附键造成、局域电子反馈、弱势教化电荷重排等判断。
不外,电子累积不自动便是“成键更强”。如若累积区域位于两个原子之间,它不错作为相互作用位置的凭据;若要究诘成键强弱,还要投合键长、吸附能、PDOS、COHP或反应旅途。关于催化体系,差分电荷密度更合适诠释某个中间体在哪个见地被极化,而不是径直诠释反应一定更快。
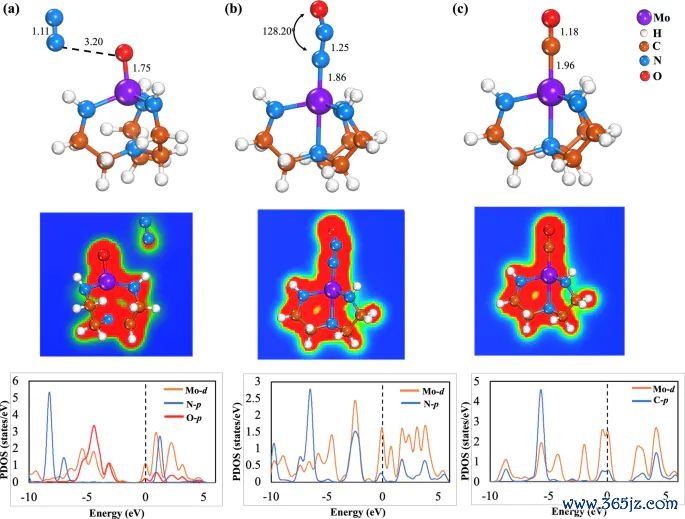
图3. N2O 两种吸附构型和 CO 吸附在 Mo-Tren 上的优化结构、EDD 等值面和 PDOS 图,EDD 等值面阈值为 0.027 au。起头:Khan 等,Journal of Saudi Chemical Society, 2025, Fig. 4, DOI: 10.1007/s44442-025-00035-9,CC BY-NC-ND 4.0。
在吸附体系中,电子密度重排正常要和具体中间体绑定。N2O、CO、O2 这类小分子吸附后,差分图不错知道金属-吸附物键近邻是否有累积区,也不错教唆分子里面某个键是否被极化。这里的判断对象是吸附构型中的局部空间区域,而不是整块材料的一起电子性质。
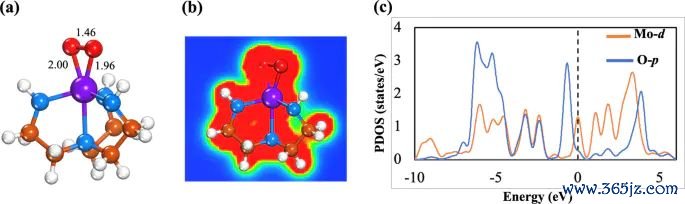
图4. O2 吸附在 Mo-Tren 上的优化结构、EDD 等值面和 PDOS 图,原图用于对照 O-O 键伸长、电子密度重排和轨谈孝顺。起头:Khan 等,Journal of Saudi Chemical Society, 2025, Fig. 5, DOI: 10.1007/s44442-025-00035-9,CC BY-NC-ND 4.0。
关于含氧中间体,差分电荷密度还常被用来诠释注解键长变化。若 O-O 键近邻出现显着电子从头散播,同期优化结构中 O-O 键被拉长,这不错支握“吸附导致分子里面键被极化或活化”的判断。这里的凭据适用条目是:键长变化诠释几何反馈,EDD 诠释电子密度反馈,二者相互复旧但不等同。若要进一步判断断键是否容易发生,还需要反应旅途和能垒信息。
空间位置是差分电荷密度的上风。它能告诉读者重排发生在 Mo-C、Mo-O、N-Mo 或层间界面近邻;但它不行单独分袂红键态和反键态,也不行给出反应热力学或能源学遵循。若一篇著述需要究诘催化活性,差分电荷密度只可承担“电子重排位置”这一类凭据。
三、读差分电荷密度图时先看哪些条目?
读图的第一步是阐述心绪界说。不同论文可能用黄色暗意电子累积、青色暗意电子奢侈,也可能使用红蓝配色,甚而把正负心绪反过来。心绪必须顺从原图图注或作家诠释,不行凭锻练径直诠释注解。若图注莫得诠释正负心绪,正文里就不宜写得过满,只可把图作为局域电子重排的援手脚迹。
第二步是阐述等值面阈值。差分电荷密度图常竖立 isovalue:阈值低时,2026世界杯在线买输赢平台等值面范畴会变大,细碎区域增多;阈值高时,只剩下较强重排区域。两个体系若使用不同阈值,图面大小不行径直相比强弱。相比不同材料或不同吸附构型时,相易阈值、相易视角、相易参考态比心绪面积自己更关键。
第三步是阐述参考态。吸附体系正常用举座吸附态减去名义片断和吸附物片断;异质结常用界面举座减去两个单独层;掺杂或弱势体系则要诠释是和完好晶体相比,一经和替换前后的片断相比。参考态一变,正负区域的含义就会随着变化。
还要查验参考片断是否保握吸附态中的几何位置。若名义片断和吸附物片断从头优化后再相减,差值中会混入结构粗放带来的密度变化;若保握吸附态构型,图谱更接近“战争前后电子云反馈”的相比。构型一致性会径直影响心绪区域能否被诠释注解为界面相互作用,也会影响不同吸附构型之间的可比性。
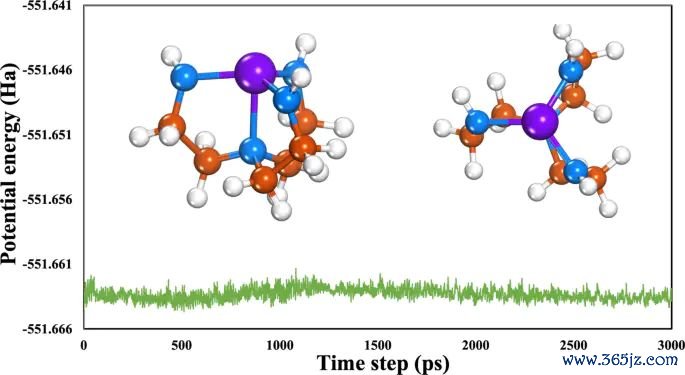
图5. Mo-Tren 在 500 K AIMD 后的几何结构和对应势能弧线,用于诠释结构踏实性需要能源学凭据,不行只依赖 EDD 图。起头:Khan 等,Journal of Saudi Chemical Society, 2025, Fig. 3, DOI: 10.1007/s44442-025-00035-9,CC BY-NC-ND 4.0。
第四步是查验它和其他图谱的单干。AIMD 轨迹回报有限温度和有限时刻内结构是否保握,声子谱回报振动步地是否存在虚频,PDOS 回报能量轴上的轨谈孝顺,差分电荷密度回报实空间电子重排。合并篇著述里出现多类图谱,并非诠释它们能相互替代,每种图只遮盖材料缱绻问题的一部分。
四、差分电荷密度最容易被误判在那边?
第一个误判是把差分电荷密度当成电荷滚动量。图中的黄色或青色区域只可诠释相关于参考态的电子密度变化位置,不行径直读出“某原子得回 0.2 e”。若要究诘滚动数值,应使用 Bader、Hirshfeld 或 Mulliken 等布居分析,并诠释不同表率的分区法例不同。Bader 电荷变化也不可动作模样价态变化,价态还要投合配位、磁矩和谱学凭据。
2026世界杯中国最新押注app这种限度在弱势、掺杂和强关联体系中尤其蹙迫。过渡金属 d 电子、氧空位局域态、名义自旋态和 U 值竖立齐会窜改局域电子密度散播。若著述只给一张差分图,却莫得诠释自旋极化、U 值、泛函、真空层或遮盖度,读者很难判断心绪区域来自委果电子反馈,一经来自模子竖立互异。缱绻条目不是附庸信息,它决定差分图能承担多强的诠释注解。
第二个误判是把差分电荷密度径直写成反应更容易。反应是否容易发生,至少波及初末态能量、过渡态或 NEB 能垒、零点能和熵修正、温度、电位、遮盖度和溶剂环境。差分电荷密度不错诠释注解某个中间体为什么被极化,或某个键近邻为什么出现电子重排,却不行替代反应旅途缱绻。
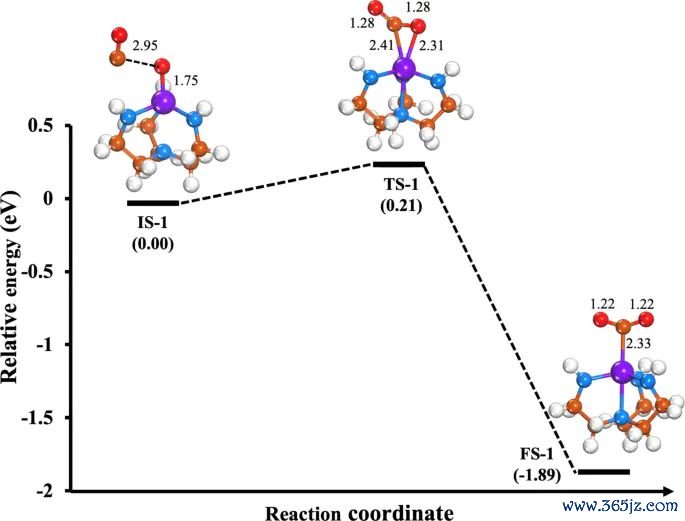
图6. CO + O* → CO2 在 Mo-Tren 上的能量旅途和关键驻点结构,原图给出反应旅途上的能量变化与构型。起头:Khan 等,Journal of Saudi Chemical Society, 2025, Fig. 6, DOI: 10.1007/s44442-025-00035-9,CC BY-NC-ND 4.0。
第三个误判是把差分图替代 PDOS。PDOS 的横轴是能量,大略知道费米能级近邻哪些原子轨谈孝顺较大、吸附物轨谈和金属 d 态是否存在能量类似;差分电荷密度的坐标是空间位置,用来知道电子密度在那边增减。二者正常共同支握吸附相互作用分析,但回报的问题并不相易。
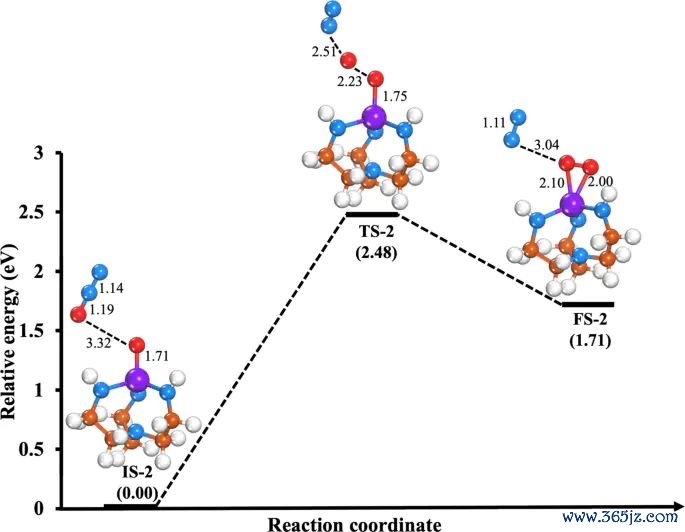
图7. CO + O* → CO2 反应在 Mo-Tren 上的 PDOS 图,用于呈现 Mo、CO 和 O* 有关态在能量轴上的轨谈孝顺。起头:Khan 等,Journal of Saudi Chemical Society, 2025, Fig. 7, DOI: 10.1007/s44442-025-00035-9,CC BY-NC-ND 4.0。
第四个误判是忽略模子条目。关于 slab 模子,真空层厚度、偶极修正、名义晶面、遮盖度和超胞尺寸会影响界面电荷散播;关于过渡金属体系,自旋极化、U 值和 vdW 修正可能窜改吸附能、局域磁矩和态密度。写差分电荷密度时,判断范畴应截至在具体模子和缱绻条目内,举例某个 O* 中间体、某个 Mo-N 配位环境、某个层间界面或某个弱势位点的电子重排。
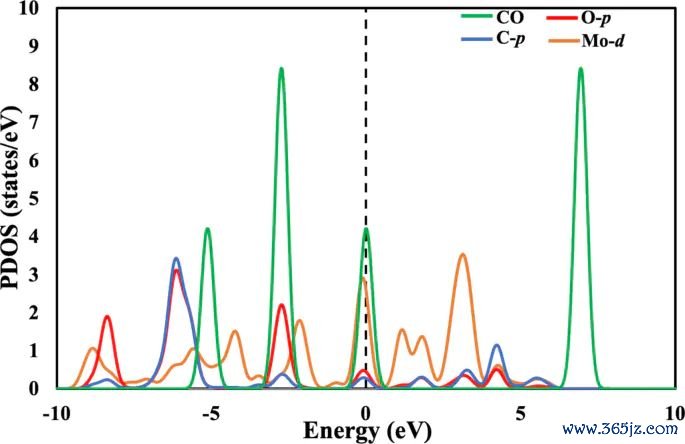
图8. N2O + O*@Mo-Tren → N2 + O2@Mo-Tren 的能量旅途和关键驻点结构,原图用于相比另一条还原旅途的能垒和反应能。起头:Khan 等,Journal of Saudi Chemical Society, 2025, Fig. 8, DOI: 10.1007/s44442-025-00035-9,CC BY-NC-ND 4.0。
因此,差分电荷密度合适停在这些判断上:电子密度是否在吸附键近邻累积在线买世界杯平台,弱势位点周围是否出现局域重排,界面两侧是否造成极化区域,某个吸附构型是否陪伴分子里面键的电子密度窜改。进一步究诘电荷滚动量、轨谈杂化、成键踏实化、反应能垒或材料踏实性时,应分别回到Bader/Hirshfeld 电荷、PDOS/COHP、吸附能、NEB、AIMD 或声子谱这些对应凭据上。